
Module 2 – Cancer Biology & Targeted Therapies
Learning
objectives
·
Awareness of
the hallmarks of cancer
·
Following
the principles of cytotoxics and combination
chemotherapy
·
Understanding
biomarkers and druggable targets
·
Targeted
therapies in the clinic
Hallmarks
of Cancer
If there is
one paper you should read to learn about Oncology, it would be Hanahan & Weinberg’s Hallmarks of Cancer paper written at the beginning of the
millennium. It has been updated in 2011 but the original paper has a certain purity
and simplicity to it.
The authors
explain that there is a set of mutations from a normal cell phenotype that are
characteristic of cancer cells. The mutations (or hallmarks) may occur in
different mutations may occur in different orders in different cells, but they
are common characteristics of a range of over 100 distinct cancer types.
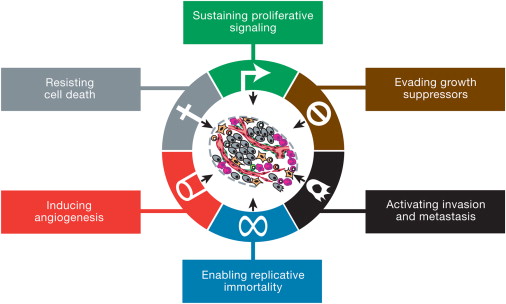
This is the
original representation of the hallmarks from the 2000 paper. They did not
include genomic instability in the original list, but argues that it was a
prerequisite for the other hallmarks to occur.
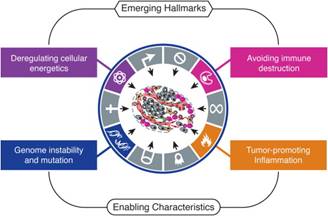
The 2011
paper updated the hallmarks to include new areas of research, considering tumour metabolism, cancer immunology, and the role of
inflammation as important hallmarks of cancer. They also upgraded Genomic
instability to a hallmark in its own right.
If you are
unfamiliar with these concepts in cancer biology, it would be worth brushing up
your knowledge before proceeding with the module by reading the hallmarks paper.
I also recommend the excellent CancerQuest website for a brief and current overview of
cancer biology.
Cytotoxic
chemotherapy
Paul Ehrlich
first coined the term chemotherapy as the process of curing illness with
chemical compounds. In his time, he was mainly referring to infection, but
these days the term is used mostly for cancer therapy. Cytotoxic chemotherapy
has a shady history, originating from mustard gas used in World War II.
Accidental exposure of Italian soldiers revealed that they had depleted white
cell counts, leading to its first clinical use in leukaemia.
Chemotherapy
is big business, with leadings drugs still bringing in billions of dollars in
sales to Pharma. Also chemotherapy is not going to be
replaced by targeted therapies, in the same way that surgery has not been
replaced by chemotherapy. A detailed review of cytotoxic chemotherapy is
available in extension module E1. The key principles to understand are as
follows:
Chemotherapy discovery. Cytotoxics are discovered through serendipity,
screening programmes, or by copying and modifying
existing drugs to enhance their bioavailability. Certain promising cytotoxics may be selected for clinical because they
demonstrate efficacy in specific tumour types, but
their action is not specific to a tumour type.
Chemotherapy mechanism. Most cytotoxics work by inhibiting cell division, either by
interfering with DNA synthesis, or damaging DNA which results in replicative
arrest, or interfering with mitosis.
Chemotherapy toxicity.
Chemotherapy
has the most dramatic effect on rapidly proliferating tissues. This is why it
affects the bone marrow and the gut mucosa. Mucositis
and neutropaenia are therefore common side effects. Nausea
is caused by the effect of chemotherapy on the area postrema
of the hypothalamus, and mediated through 5HT-3 and neurokinin
receptors. We now have very effective anti-emetics that can block these
pathways.
Chemotherapy scheduling. We give cytotoxics
in cycles, typically every 3 weeks. The reason for this is to allow the bone
marrow to recover from the effects of chemotherapy. High-dose chemotherapy is
used in the treatment of haaematological malignancy
to ablate the bone marrow, because this is the origin of the malignancy. The
patient has to be ‘rescued’ by a bone marrow transplant.
Combination chemotherapy. We often use chemotherapy in combinations.
There may be mechanistic synergy between the different agents (e.g. drugs
targeting cells at different points in the cell cycle), and using drugs in
combination means that the tumour is less likely to
become resistant to therapy. It also means we can continue treatment if a
patient develops a dose-limiting toxicity from one of the drugs.
If you can, try and observe the chemotherapy
nurses as they discuss chemotherapy with a patient starting on treatment.
Druggable targets
Modern
translational oncology follows a new type of path towards the development of
blockbusting new drugs:
·
Build a
low-level model of your tumour, using cancer genetics
and in-vitro / animal models.
·
Identify key
pathways that seem to be dysregulated in this tumour (canonical pathways)
·
Identify the
driver mutations in these pathways and work out what these do in the cell,
usually at the protein level (a biomarker of this disease).
·
Build a
compound to target these mutations, usually by blocking a receptor.
·
At the same
time, build a test (biomarker assay) to tell whether a tumour
has this mutation or not, and thereby predict whether
or not the drug is likely to work for a given patient.
·
Test your
drug for efficacy and side effects (so-called off target effects) because no
compound is 100% specific to your chosen cancer target, and many pathways may
not be 100% specific to cancer cells.
What is
exciting in oncology is that this process has been tremendously successful for
some conditions where we previously did not have very good treatments. In this
diagram, we map the new biological therapies onto the hallmarks of cancer, so
you can see how much of our understanding of cancer biology has been turned
into potential cancer therapy:
|
|
|
Figure 1: Druggable targets and their clinical status, overlaid
onto the Hanahn & Weinberg hallmarks of cancer |
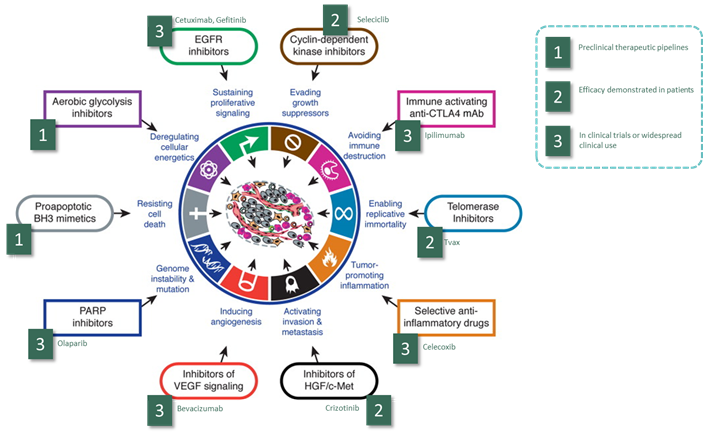
Unlike
chemotherapy agents, the biological therapies tend to have similar yet
unpronounceable names. However there is a method to the madness – and if you
know the code, it can make you sound very clever indeed. For me it’s a bit like
the day that someone explained to me that all bacteria ending in –ella are gram negative.
|
Name
element |
Meaning |
Example |
|
-mab |
Monoclonal antibody |
Trastuzumab |
|
-ib |
Small molecule inhibitor |
Erlotinib |
|
-ximab |
Chimeric human-mouse antibody |
Cetuximab |
|
-zumab |
Humanised mouse antibody |
Pertuzumab |
|
-ci- |
Circulating system target |
Bevacizumab |
|
-tu- |
Tumour target |
Cetuximab |
|
-tin- |
Tyrosine kinase inhibitor |
Afatinib |
|
-zom- |
Proteasome inhibitor |
Bortezomib |
So for
example the generic name of Herceptin is Trastuzumab.
We therefore know that the drug binds a tumour target
and is a humanized mosu antibody therapy. In contrast
Gleevec or Imatinib (the
wonder drug for CML and gastrointestinal stromal tumours)
is a small molecule inhibitor of tyrosine kinase. Go ahead and dazzle your
friends with your newfound knowledge!
Targeted
therapies in the clinic
As I
mentioned above we have some real success stories for novel targeted therapies
in the clinic, and you might meet patients on these therapies during your attachment.
We will drill down into four different examples:
·
B-RAF Kinase
inhibitors in melanoma
·
Targeted
cancer immunotherapy
·
EGFR and ALK
inhibitors in non-small cell lung cancer
·
Anti-angiogenic therapy in Neurofibormatosis
Type 2
B-RAF
kinase inhibitors in melanoma
The B-RAF
gene is a classic example of a proto-oncogene. It encodes an intracellular
signal transduction protein belonging to the RAF family, and falls in the MAP
kinase pathway. Mutation of the B-RAF gene to become constitutively active is
found in a range of common cancers. Approximately 40-60% of patients with
melanoma will have a specific mutation in the B-RAF gene (V600E).
|
|
|
Figure 2:
Diagram illustrating key regulator effects mediated by BRAF protein |
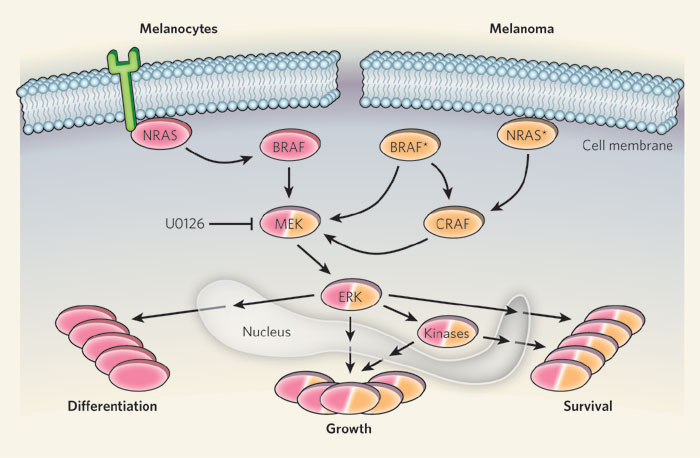
Why is this important?
Well up until about 5 years ago, we had very little in the way of effective
therapies for melanoma. Dacarbazine, a very old
alkylating agent, used to be used for fit patients. It is very toxic and has
poor response rates (it works in about 5-8% of patients). Now we have novel
targeted therapies which can inhibit the mutated gene product. As a result the drugs and high specificity and much fewer
off-target effects, because the mutated protein is only found in tumour cells. Agents such as Sorafenib
and Vemurafenib have a response rate of 50-60%, and
are generally very well tolerated oral therapies. It is fair to say that the
availability of these new drugs has transformed the outlook for patients with
advanced melanoma.
Targeted
cancer immunotherapy
Over the
last decade the understanding of the role of the immune system in
carcinogenesis, and specifically the mechanisms by which tumour
cells evade detection and eradiation by the immune system has been studied,
leading to the discovery of new therapies. The programmed cell death receptor
is catchily abbreviated to PD-1 (I would have called
in DEATH-1 if I discovered it) and is an important immune surveillance checkpoint
involved in self-tolerance. PD-1 is overexpressed in a range of tumours, including melanoma, and many tumour
cells will also overexpress PD-L1 which is the ligand to PD1. This is a good
example of an autocrine loop where tumour cells will produce a signal that promotes its own
proliferation or survival.
|
|
|
Figure 3 :
Nivolumab is an antibody that disrupts the
inhibitory signals that prevent T-cells from recognizing the tumour cells as being abnormal |
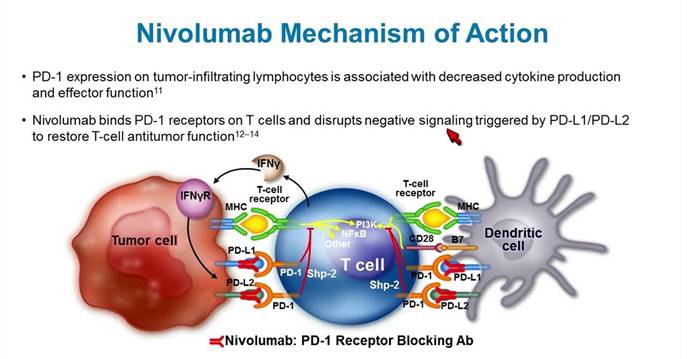
Pembrolizumab and Nivolumab are
monoclonal antibody therapies that inhibit the PD-1 receptor, which prevents
the ability of tumour cells from evading the immune
system. The treatment is quite toxic, with a significant risk of severe
hepatitis, but can often induce long lasting remissions of tumours.
Ipilimumab is another antibody therapy directed at the
CTLA4 receptor which is a similar fashion has an inhibitory effect on cytotoxic
T-lymphocytes.
The
magnitude of effect of nivolumab in combination with ipilimumab to activate the immune system is quite
incredible. In 2015 the Checkmate study was published showing clinical outcomes
for patients with advanced melanoma treated with this combination. Response
rates were as high as 60% and further in those patients that responded, some had complete remission of their metastatic disease
for over a year. It even caught the attention of the press (click here for BBC article). The downside – well these drugs are
extremely expensive – ipilimumab costs $120,000 for 4
treatments and Nivolumab costs $25,000 for 4
treatments, and they can be extremely toxic too.
EGFR and
ALK inhibitors in non-small cell lung cancer
The treatment
landscape for metastatic non-small cell lung cancer (NSCLC) has also changed
tremendously in the last few years. This group of patients often had
significant cardiovascular and respiratory co-morbidities, and treatment with cisplatin containing chemotherapy was quite toxic.
Activating
EGFR mutations occur in between 10 and 30% of NSCLC patients, depending on
ethnicity and smoking status. Higher mutations rates are observed in
non-smokers and patients with adenocarcinoma histology. Ethnicity is incredibly important. One of the
EGFR inhibitors was initially developed and trialled
in Japan, where the prevalence of EGFR mutations and adenocarcinoma histology
is nearly 50%. The company then wanted to try the drug in the US, where EGFR
mutation rates are much lower (5-10%). The problem is that in these first
studies, they did not stratify patients to therapy according to EGFR status.
Nowadays, EGFR testing is performed routinely on patients with NSCLC,
particularly adenocarcinoma subtype and they are a predictive and prognostic
assay. This means they both predict a response to therapy and act as an
independent factor in patient survival. Drugs such as Erlotinib
and Gefinitib are examples of oral EGFR tyrosine
kinase inhibitors that have been used with great success in the clinic.
|
|
|
Figure 4 :
Importance of EGFR and ALK pathways, important in non-small cell lung cancer |
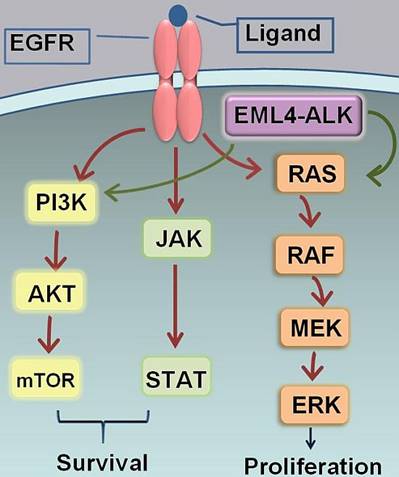
ALK is a
similar fusion oncogene that has an activating effect on cell proliferation. It
is important because some EGFR negative tumours will
have an ALK mutation, and this represents a druggable
target. New small molecule tyrosine kinase inhibitors such as crizotinib and ceritinib target
this pathway and may provide a treatment strategy for patients who do not
harbor EGFR and ALK mutations.
If you go to
lung cancer clinic, it’s worth asking about treatment for EGFR and ALK positive
patients as this is a rapidly evolving field (that’s a polite way of saying my
crib notes might be out of date already!)
cKIT inhibition in myeloid leukaemia
and GIST
Imatinib (Glivec) is a
really good example of a targeted therapy ‘success story’ and was one of the
first targeted therapies to come into common ptractice.
It targets an oncogenic fusion protein called BCR-ABL, which forms a constitutively
active tyrosine Kinase. The BCR-ABL translocation is also known as the
Philadelphia chromosome, a mutation found in some chronic myeloid leukaemia which used to be resistant to therapy.
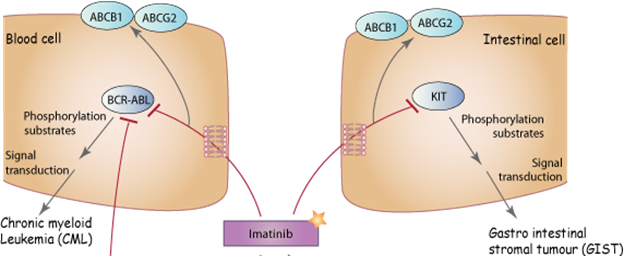
The same
drug also targets cKIT which is overexpressed in a
range of tumours, including melanoma, seminoma and a
rare condition known as gastro-intestinal stromal tumour
or GIST. Although GIST are rare, they are quite common
in the Oncology Centre because they are an area of clinical interest for some
of our oncologists! GISTS are hard to treat with conventional chemotherapy and
radiotherapy, because the rich layer of stromal cells protects them from these
treatments. Glivec can yield really dramatic
responses in GIST, but you need to know how to look for the signs of a
response, because responding tumours do not always
shrink
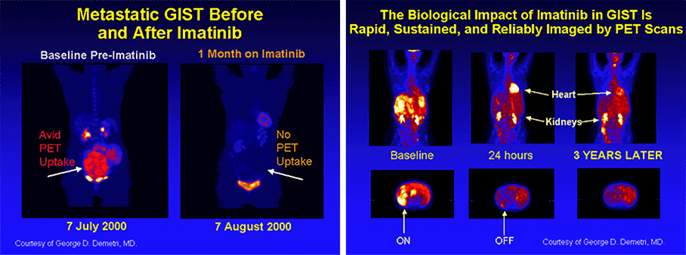
FDG PET
scanning reveals the reduction in metabolic activity, which can be seen very
quickly after starting therapy.
Treatment
assessment and RECIST criteria
This brings
us to an interesting point – how do we assess disease response in solid tumours? In most cases radiologists use the RECIST criteria
to report treatment response on cross sectional imaging. It’s worth knowing a
little about this. RECIST stands for Response Evaluation Criteria in Solid
Tumors. They are a standardized method of assessing tumour
response used in clinical trials. A radiologist identifies an index lesion that
will be evaluated during treatment, and perform linear measurements of the
lesion. Response criteria are evaluated as
·
CR (complete
response) = disappearance of all target lesions
·
PR (partial
response) = 30% decrease in the sum of the longest diameter of target lesions
·
PD
(progressive disease) = 20% increase in the sum of the longest diameter of
target lesions
·
SD (stable
disease) = small changes that do not meet above criteria
In other
conditions such as germ cell tumours, advanced
prostate cancer or ovarian cancer, we use a serum tumour
marker (HCG & AFP, PSA and CA12.5 respectively)to
assess treatment response. It’s worth remembering that serum markers may drop
before radiological changes are observed in a tumour.
It’s also
worth remembering that some tumours can apparently
increase in size as a response to treatment (pseudoprogression),
and sometimes in the case of metastatic disease, some leasions
can shrink in response to therapy whilst others increase in size (differential
response), reflecting differences in the underlying biology of different sites
of metastatic disease.
There is a
lot of interest in being able to use circulating tumour
DNA as a
marker of tumour load, and this is a strong research
focus in the CRUK Cambridge Institute at present. CancerGRACE
has a great video explaining the technique.
Resistance
to therapy
Wonderful as
these targeted therapies may sound, they do not work forever, and eventually resistance
develops. The precise lock-and key nature of these agents makes them prone to ‘steric
hindrance’, that is a small mutation in the proteins responsible for the shape
of a receptor that prevent a TKI or antibody from
binding to the target:
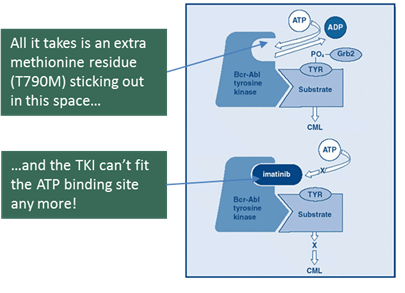
Most
targeted therapies will work for a period of 10-12 months, and then it becomes
necessary to switch to another agent. We are fortunate that is a range of tumours, there are several agents now available that target
these pathways, allowing for rotation of treatment when resistance develops.
Conclusion
Systemic
therapies in oncology is a huge
area. I hope this module gives you some insight into the way that we have come
from a background of using non-targeted cytotoxic drugs and are now moving
towards a more mechanistic approach to cancer therapy. If you see a chemotherapy
patient in clinic, take a look at their treatment history to try and understand
the decision making behind their choice of treatment.
If
you want to take a deeper dive into the world of chemotherapy, take a look at
Module E1 on chemotherapy and Biological therapies.